UniGuide: Unified Guidance for Geometry-Conditioned Molecular Generation
Johanna Sommer
The field of computational biology and drug discovery has witnessed a revolutionary breakthrough with the development of AlphaFold, an AI system that has transformed our understanding of protein structures. This groundbreaking achievement was recently recognized with the 2024 Nobel Prize in Chemistry, awarded to Demis Hassabis and John Jumper of Google DeepMind, along with David Baker of the University of Washington. Building on this success, AlphaFold 3 has taken a significant leap forward by incorporating diffusion-based generative models into its architecture. This latest iteration of the model employs a novel approach that directly models the generation of atomic coordinates, allowing for highly accurate predictions of bio-molecular interactions across a wide spectrum of molecular types. This advancement has not only expanded the system's capabilities beyond proteins but has also set new benchmarks in the field of drug discovery and design.
Besides protein structures, molecular geometries play a fundamental role in drug discovery, directly impacting how molecules interact with their biological targets. With the rise of deep generative models like diffusion models, which excel at modeling and generating molecular geometries across all aspects of the discovery process, machine learning has become central to modern drug discovery. Traditionally, tailoring molecules to specific geometric constraints, such as binding to a protein pocket, required specialized generative models. We introduce UniGuide, a framework that integrates diverse geometric conditions into molecular generation, enabling adaptability without additional training or external networks. In doing so, UniGuide represents a significant step toward a universal solution for drug design challenges.


Why do we need a condition map?
At the core of UniGuide lies its condition map—a transformative function that translates complex geometric constraints into a space that is compatible with molecular geometries. Unlike traditional approaches that require either re-training models for each specific task or additional proxy models, UniGuide takes advantage of off-the-shelf, unconditional molecular generators, enabling flexible guidance without retraining. This plug-and-play framework, depicted in Fig. 1, can tackle various challenges, including:
- Structure-Based Drug Design (SBDD): Designing molecules that bind to a given protein pocket. When a molecule binds well to a protein, it can regulate its activity or perform therapeutic interventions.
- Fragment-Based Drug Design (FBDD): Expanding molecules based on known fragments or fragment sets. Often, it is known that a certain molecular fragment interacts well with a specific binding site and chemists then iteratively add new chemical groups to improve the fragment’s affinity, specificity, and drug-like properties.
- Ligand-Based Drug Design (LBDD): Generating molecules that are in accordance with predefined 3D volumes. Similarly to SBDD, LBDD aims to generate molecules that bind well to a given protein. It is, however, difficult to obtain high-quality data in the field of drug discovery, and this specifically holds for protein data. As a consequence, in this task, we try to match the 3D volume of a ligand, of which we know that it has good binding affinity - without using information about the protein.

How is the condition map used at generation time?
At inference time, the condition map plays a crucial role in guiding the generation process of diffusion models, ensuring that generated outputs meet desired geometric conditions. In our framework, self-guidance leverages this condition map to provide a unified and adaptable mechanism for molecular generation tasks. The condition map translates arbitrary geometric constraints, such as shapes, densities, or structural templates, into a form compatible with the generative process. This transformation aligns the target condition (derived from the input constraint) with the representation space of the diffusion model, see Fig. 2.
Diffusion models for molecular geometries operate by gradually transforming structured molecular data into random noise through a forward process and learning the reverse process to reconstruct structured data.
The forward diffusion step involves progressively adding Gaussian noise to a molecule’s atomic coordinates and features, incrementally degrading the structured geometry into a state of randomness. This gradual addition of noise is carefully controlled by a predefined noise schedule, ensuring that each step maintains a balance between introducing sufficient variability and preserving essential structural information.
During training, the model learns to reverse this process by predicting and removing noise at each step. This reverse diffusion process is guided by a neural network, often leveraging graph neural networks (GNNs). GNNs process the molecular graph by iteratively passing and aggregating information between atoms, effectively capturing both local and global interactions. By incorporating spatial features such as inter-atomic distances, GNNs ensure that the three-dimensional geometry of the molecule is accurately represented and maintained throughout the diffusion process. Additionally, Equivariant GNNs are employed to preserve the molecule’s properties under rotations and translations, ensuring that the generated geometries are physically and chemically consistent. This GNN is consequently trained to predict and remove the added noise at each step. During training, the network is exposed to various noisy states of the molecule and learns the patterns that distinguish meaningful chemical structures from random noise. By iterative denoising, the model refines the atomic positions and features, effectively “sculpting” the molecule back to its original, stable configuration. The model thus learns the probability distribution of molecular geometries, enabling it to generate realistic molecules starting from pure noise during inference.
Self-guidance in diffusion models works by modifying the reverse diffusion process. During this process, the model predicts noise at each step and adjusts it iteratively to approach the target condition. By integrating the condition map, UniGuide calculates a guidance signal that steers the generation toward meeting the desired constraints. For instance, when generating a molecular structure that conforms to a specific shape, the condition map aligns the molecule’s generated conformation with the given shape while preserving the model’s inherent generative capabilities. The self-guidance mechanism adapts the noise prediction to include the distance between the generated data and the condition, see Fig. 2. This distance quantifies how well the generated structure matches the condition. The model then incorporates this information as a corrective signal, iteratively refining the generated structure. Importantly, the process retains the unconditional model’s generality and flexibility, allowing UniGuide to adapt to various tasks by simply adjusting the condition map.
Why does this matter?
UniGuide is able to address critical challenges in drug discovery by combining adaptability, efficiency, and performance in a single, cohesive framework. Unlike traditional task-specific models that require dedicated training for each objective, UniGuide’s unified approach allows it to tackle a wide range of tasks, from binding-site-specific ligand design to scaffold elaboration and fragment assembly. This adaptability eliminates the need for multiple specialized systems, streamlining workflows and providing researchers with a flexible tool capable of handling diverse geometric constraints.
By decoupling training from task-specific conditions, UniGuide significantly reduces computational overhead. Instead of training new models or modifying existing architectures for each application, researchers can use a single unconditional diffusion model across scenarios, saving both time and resources. This efficiency makes UniGuide accessible even to labs with limited computational capabilities.
Most importantly, UniGuide achieves these benefits without compromising performance. Across benchmarks for ligand-based, structure-based, and fragment-based drug design tasks, it consistently matches or surpasses specialized models, delivering competitive docking scores, superior shape alignment, and effective fragment linking while maintaining chemical diversity.
How does it compare to specialized models?
UniGuide’s versatility can be seen in its broad spectrum of applications across various domains of molecular generation.
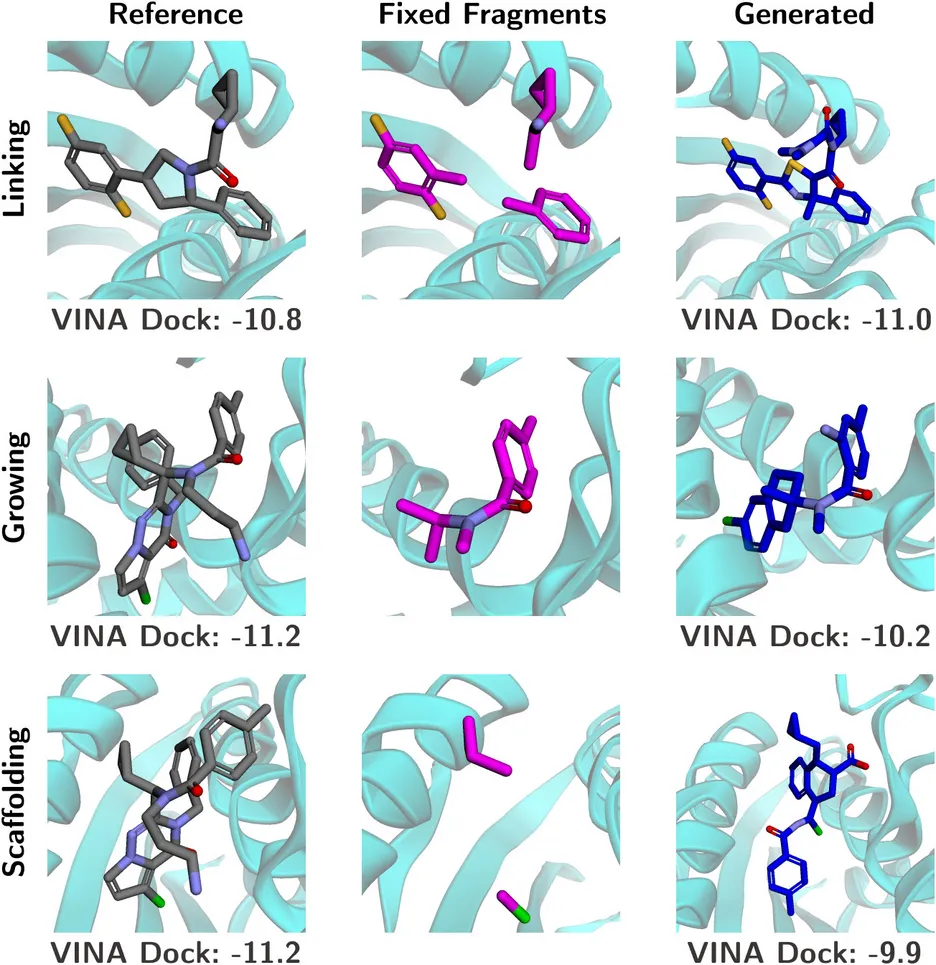
For Fragment-Based Drug Design (FBDD), UniGuide can address multiple sub-tasks, leveraging its condition map framework to tackle specific challenges, see Fig. 3. While such subtasks have different objectives like changing the core structure of a molecule, combining multiple active components or adding additional functional groups, they can be abstracted to the same underlying methodology - completing a molecule given a partial molecular geometry. As a consequence, UniGuide can tackle this wide range of tasks with the same underlying condition map.
- Linker Design: UniGuide generates molecular linkers to connect predefined fragments, ensuring geometric alignment and chemical validity. This task is critical when fragments are spatially separated, and the generated linkers must bridge them effectively while maintaining drug-like properties.
- Fragment Growth: Expanding molecular scaffolds, UniGuide adds functional groups or chemical structures to enhance binding affinity (VINA scores) or explore new chemical spaces. The framework adapts to specific growth directions, preserving the scaffold’s structural integrity and pharmacological relevance.
- Scaffold Hopping: UniGuide facilitates replacing molecular cores while retaining functional group interactions.
- Functional Group Optimization: For fragments with moderate binding affinity, UniGuide can fine-tune specific regions to, in the future, enhance properties like solubility, stability, or receptor interactions.
- Multi-Fragment Assembly: UniGuide combines multiple fragments into cohesive molecular structures, ensuring spatial and chemical harmony.
For Ligand-Based Drug Design (LBDD), UniGuide can generate molecules that achieve state-of-the-art shape similarity to reference ligands. These molecules not only align closely with the predefined 3D geometries but also maintain chemical diversity, see Fig. 4. An off-the-shelf unconditional model can now be modified with a few steps to design molecules with a specific 3D volume. UniGuide consistently outperforms specialized models in key metrics, such as the balance between molecular shape alignment and graph dissimilarity, showcasing its potential as a more performant alternative to task-specific approaches.
For Structure-Based Drug Design (SBDD), UniGuide showcased its ability to harmonize ligand-protein interactions effectively. By guiding the generative process of the protein pocket, UniGuide produces ligands that exhibit competitive docking scores (VINA scores). The VINA score is an important metric used in molecular docking to predict and quantify the binding affinity between a ligand and its target protein. This computational assessment allows for the identification of those molecules that are most likely to bind effectively to their targets. Consequently, we can use the VINA score to measure the quality of our generated ligands in terms of binding affinity. Importantly, at times UniGuide even improves over the docking scores achieved in the dataset. This performance highlights UniGuide’s adaptability, as it can operate on both protein and ligand levels without requiring additional training.
Having demonstrated UniGuide’s competitive performance across a wide range of established tasks and benchmarks, we now highlight its plug-and-play capability for tackling new challenges. For example, when targeting a protein pocket, additional information about specific regions of interest—such as the placement of certain atom types in designated locations to enhance binding affinity—can be crucial. Existing specialized models would require designing new embeddings, modifying the architecture, and incurring significant training overhead, along with the need for extensive high-quality training data. In contrast, UniGuide simplifies this process by prompting researchers to only create a condition map that directs the generative process to position specific atom types in the regions of interest. This approach leverages existing trained models, enabling the efficient adaptation to new tasks without the need for substantial architectural changes or additional data.

A Unified Framework for Molecular Generation
UniGuide takes a step toward a unified approach to molecular generation. By bridging the gap between task-specific models, it not only accelerates drug discovery but also sets a foundation for future innovations in molecular design, both in terms of novel tasks that can now be tackled and novel generative models that now become readily available for downstream tasks. As the field evolves, frameworks like UniGuide promise to streamline the landscape of AI-guided molecular design.