Diffusion Models for Rigid Protein-Protein Docking and Binding Pocket Conditioned Receptor Flexibility
Results of this project are explained in the final report.

- Sponsored by: Massachusetts Institute of Technology (MIT) and TUM Chair of Bioinformatics I12
- Project Lead: Dr. Ricardo Acevedo Cabra
- Scientific Lead: PhD candidate Céline Marquet and PhD candidate Hannes Stärk
- TUM Co-Mentor: Dr. Ricardo Acevedo Cabra
- Term: Winter semester 2022
Goal
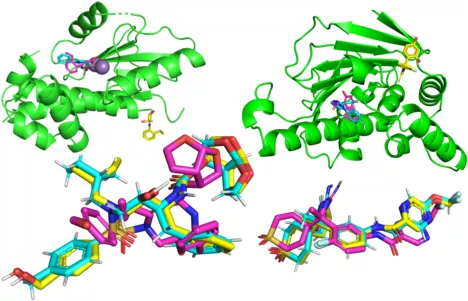
A central challenge of drug discovery is identifying drug candidates that bind to and interact with a target protein [1]–[3]. Such target proteins could, e.g., be penicillin-binding proteins within bacteria, where the protein-drug binding mechanism indirectly leads to the bursting of bacteria [4]. In most cases, databases of known drug candidates are scanned to find possible targets [3], which limits the solution space to already known candidates. Our goal is to circumvent this limitation and make drug discovery more efficient by generating molecules de novo with a binding-pocket conditioned diffusion model taking the 3D structure and binding site of a target protein as input.
Main Methods:
This project is based on several geometric deep learning methods, such as Graph Neural Networks (GNNs) [5]. We aim to use denoising diffusion probabilistic models, proposed in Ho et al., in 3D euclidean space to generate desired drug molecules [6], [7]. Hoogebom et al. previously implemented this approach for molecule generation without specific conditioning on a target protein binding pocket [8]. We will build on this work, and additionally condition the denoising model on the 3D structure of a protein.
To achieve this, we will explore two different approaches:
- Summarizing the protein’s binding pocket via an E3 Equivariant Graph Neural Network [9] and including it as input to the denoising model.
- Performing the denoising process inside of the protein’s binding pocket with it being informed by the distances between the molecule and protein atoms.
In brief, this geometric deep learning project includes methods such as:
- Graph Neural Networks
- Denoising diffusion models [6], [7]
- E3 Equivariant graph neural networks [9]
Data:
To train our model we will rely on the commonly used dataset PDBBind [10], which is a subset of the Protein Data Bank (PDB) [11] that provides 3D structures of individual proteins and complexes. PDBBind v2020, contains 19 443 protein-ligand complexes with 3890 unique receptors and 15 193 unique ligands. Furthermore, to evaluate the generalization capabilities of our models, we will use the time split provided by [12] as well as a scaffold split and a protein sequence similarity-based split.
Accepted students to this project should attend (unless they have proven knowledge) online workshops at the LRZ from TBA. More information will be provided to students accepted to this project.
References
[1] R. Santos et al., “A comprehensive map of molecular drug targets,” Nat. Rev. Drug Discov., vol. 16, no. 1, Art. no. 1, Jan. 2017, doi: 10.1038/nrd.2016.230.
[2] J. Ha, H. Park, J. Park, and S. B. Park, “Recent advances in identifying protein targets in drug discovery,” Cell Chem. Biol., vol. 28, no. 3, pp. 394–423, Mar. 2021, doi: 10.1016/j.chembiol.2020.12.001.
[3] J. Hughes, S. Rees, S. Kalindjian, and K. Philpott, “Principles of early drug discovery,” Br. J. Pharmacol., vol. 162, no. 6, pp. 1239–1249, Mar. 2011, doi: 10.1111/j.1476-5381.2010.01127.x.
[4] P. Macheboeuf, C. Contreras-Martel, V. Job, O. Dideberg, and A. Dessen, “Penicillin Binding Proteins: key players in bacterial cell cycle and drug resistance processes,” FEMS Microbiol. Rev., vol. 30, no. 5, pp. 673–691, Sep. 2006, doi: 10.1111/j.1574-6976.2006.00024.x.
[5] F. Scarselli, M. Gori, A. C. Tsoi, M. Hagenbuchner, and G. Monfardini, “The Graph Neural Network Model,” IEEE Trans. Neural Netw., vol. 20, no. 1, pp. 61–80, Jan. 2009, doi: 10.1109/TNN.2008.2005605.
[10] Z. Liu et al., “Forging the Basis for Developing Protein-Ligand Interaction Scoring Functions,” Acc. Chem. Res., vol. 50, no. 2, pp. 302–309, Feb. 2017, doi: 10.1021/acs.accounts.6b00491.
[11] H. Berman, K. Henrick, and H. Nakamura, “Announcing the worldwide Protein Data Bank,” Nat. Struct. Mol. Biol., vol. 10, no. 12, Art. no. 12, Dec. 2003, doi: 10.1038/nsb1203-980.