Creating a single-cell atlas of human blood
Results of this project are explained in the final report.
- Sponsored by: TUM Chair of Mathematical Modeling of Biological Systems (MDSI Prof. Theis) and Institute of Computational Biology (Helmholtz Zentrum München)
- Project Lead: Dr. Ricardo Acevedo Cabra
- Scientific Lead: PhD candidate Karin Hrovatin, Dr. Malte Luecken, PhD candidate Christopher Lance, PhD candidate Lisa Sikkema
- TUM Co-Mentor: Prof. Massimo Fornasier
- Term: Winter semester 2022
Motivation
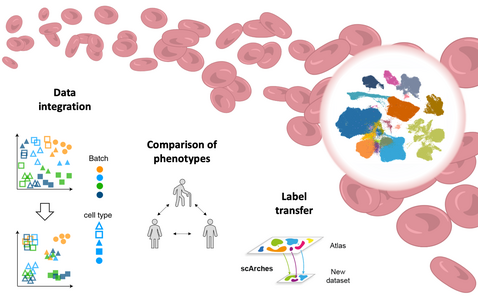
Cells are the fundamental building blocks of tissues, organs and entire organisms. Breakthrough technologies for profiling RNA in individual cells (scRNA-seq) provide new insights into the functioning of and interaction between cells, which has led to a leap forward in biomedical science.1–3. While great strides have been taken forward, current single-cell datasets typically contain many cells, but only few samples, failing to represent the diversity existing in human populations. To comprehensively understand a human biological tissue in health and disease, it is therefore necessary to capture population-level diversity by aggregating data across conditions, individuals and datasets into a single “reference”4. However, the creation of cross-dataset references is complicated by technical artifacts in the data (batch effects) that arise from differences in experimental design, making direct comparison of multiple datasets impossible. To enable cross-dataset analyses, approaches that integrate data and remove batch effects are used to construct so-called “integrated atlases”5,6. Leveraging these computational advances, multiple initiatives that aim at building integrated atlases have been started7-4. However, many organs are still missing adequate atlases, including the blood, which plays a vital role in the immune system and of which a wide variety of single-cell datasets have been generated8–10. We have recently made the first attempts to integrate peripheral blood datasets into a single atlas that must be further evaluated and iteratively improved to construct a final reference atlas, as proposed below.
Goals
- Evaluate the quality of our initial PBMC atlas including single-cell RNA-sequencing (scRNA-seq) data of >8 million cells and >1500 healthy and diseased individuals from 22 different peripheral blood mononuclear cell (PBMC) datasets across different biological conditions
- Iteratively improve the PBMC reference atlas by fine-tuning of the integration protocol
- Describe the molecular heterogeneity captured within the atlas and test how it can be used to provide context to new data by transfer learning
- Speed up methods used for large scRNA-seq data analysis
Methods/tasks
- Data preprocessing: Measurements obtained from scRNA-seq are not perfect and need quality assessment from different perspectives (e.g. removal of dying cells) with existing and new metrics. Furthermore, as metadata is collected in a study-specific manner and not directly comparable, we will align different terms used across datasets in accordance with standard terminologies.
- Evaluation and improvement of integration: There is no single best way to build an integrated atlas, thus we will take effort to optimize this process by consecutive integration evaluation followed by re-integration. For example, every batch effect removal also removes part of biological variation. This trade-off can be fine tuned with integration strength and improved preprocessing and assessed by quantifying the conservation of cross-dataset effects of disease (COVID-19) and cell type identity. Furthermore, it is unclear what should be chosen as a batch variable, i.e. the variable that is expected to cause significant technical artifacts. Therefore, we will look into the batch effect per covariate such as biological samples, subsets of datasets, or whole datasets. Lastly, atlases can be built from all available data, or from a subset of datasets, such as healthy samples alone followed by transfer-learning based mapping of diseased data on top of healthy. We will compare the two approaches based on batch effect removal and biological information conservation metrics.
- Using the atlas: At the end we will explore the atlas to examine the captured biological information in terms of cell types present in the atlas and effects of external factors, such as disease, on cell function. We will contextualize new data by mapping it on top of the atlas, such as for automatic cell type and state transfer.
- Speed up clustering, visualization, and other processing steps for large tabular data and nearest-neighbors graphs from the atlas by adapting existing methods developed for working with large data or defining and testing new approaches where necessary.
Taken together, in this project we will generate a single-cell reference atlas of blood capturing the variation of a large human population. This is of tremendous value for the research community investigating blood cell states in health and disease.
Accepted students to this project should attend (unless they have proven knowledge) online workshops at the LRZ from TBA. More information will be provided to students accepted to this project.
References
1. Tang, X., Huang, Y., Lei, J., Luo, H. & Zhu, X. The single-cell sequencing: new developments and medical applications. Cell Biosci. 9, 53 (2019).
2. Lei, Y. et al. Applications of single-cell sequencing in cancer research: progress and perspectives. J. Hematol. Oncol. 14, 91 (2021).
3. Ren, X. et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell 184, 5838 (2021).
4. Sikkema, L. et al. An integrated cell atlas of the human lung in health and disease. bioRxiv 2022.03.10.483747 (2022) doi:10.1101/2022.03.10.483747.
5. Luecken, M. D. et al. Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods 19, 41–50 (2022).
6. Lotfollahi, M. et al. Mapping single-cell data to reference atlases by transfer learning. Nat. Biotechnol. 40, 121–130 (2022).
7. Ando, Y., Kwon, A. T.-J. & Shin, J. W. An era of single-cell genomics consortia. Exp. Mol. Med. 52, 1409–1418 (2020).
8. Chen, D. et al. Single-cell atlas of peripheral blood mononuclear cells from pregnant women. Clin. Transl. Med. 12, e821 (2022).
9. COvid-19 Multi-omics Blood ATlas (COMBAT) Consortium et al. A blood atlas of COVID-19 defines hallmarks of disease severity and specificity. bioRxiv (2021) doi:10.1101/2021.05.11.21256877.
10. Wilk, A. J. et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 26, 1070–1076 (2020).